Neurobiology and Neurophysiology of Down Syndrome
 Down syndrome (DS) is the most commonly diagnosed chromosomal condition and the most common genetic cause of intellectual disability in the United States. DS affects a range of behavioral domains in children, including motor and cognitive function. Defective oligodendrocyte (OL) development and myelination have been identified in DS human brains and mouse models. This finding is consistent with studies demonstrating reduced white matter integrity in individuals with DS. The Gallo and Haydar Laboratories are collaborating to answer key questions regarding the timing and source of OL dysmaturation, and particularly whether cell-autonomous or non-cell-autonomous mechanisms lead to altered cellular differentiation and myelination. Our gene, protein and bioinformatics studies of human DS brain indicate impairments in the maturation of oligodendrocyte precursor cells (OPCs) into myelinating OLs (mOL). We have also shown that these same human brain gene and protein expression changes are found in the SCWM and cerebellum of the Ts65Dn mouse model of DS, and that they are correlated with significant in vivo abnormalities in OL differentiation and myelin production. Importantly, these cellular changes are accompanied by slower action potential conduction velocity between cerebral hemispheres, indicating that this newfound cellular deficit may play a large role in intellectual disability in DS. A detailed examination of the trisomic OLs and neurons is now warranted so that therapies can be designed to prevent the hypomyelination and its effects on motor and cognitive development in DS.
Down syndrome (DS) is the most commonly diagnosed chromosomal condition and the most common genetic cause of intellectual disability in the United States. DS affects a range of behavioral domains in children, including motor and cognitive function. Defective oligodendrocyte (OL) development and myelination have been identified in DS human brains and mouse models. This finding is consistent with studies demonstrating reduced white matter integrity in individuals with DS. The Gallo and Haydar Laboratories are collaborating to answer key questions regarding the timing and source of OL dysmaturation, and particularly whether cell-autonomous or non-cell-autonomous mechanisms lead to altered cellular differentiation and myelination. Our gene, protein and bioinformatics studies of human DS brain indicate impairments in the maturation of oligodendrocyte precursor cells (OPCs) into myelinating OLs (mOL). We have also shown that these same human brain gene and protein expression changes are found in the SCWM and cerebellum of the Ts65Dn mouse model of DS, and that they are correlated with significant in vivo abnormalities in OL differentiation and myelin production. Importantly, these cellular changes are accompanied by slower action potential conduction velocity between cerebral hemispheres, indicating that this newfound cellular deficit may play a large role in intellectual disability in DS. A detailed examination of the trisomic OLs and neurons is now warranted so that therapies can be designed to prevent the hypomyelination and its effects on motor and cognitive development in DS.
In a separate project, we are investigating locomotor dysfunction in DS. Clinical assessments indicate a range of locomotor deficits in DS, as well as slower adaptive control. Longitudinal data also indicates altered gait evolution from childhood to adulthood. Cerebellar pathology has been consistently observed in DS and is thought to contribute to dysfunction in locomotor and adaptive motor skills. Studies in animal models of DS have also indicated deficient cerebellar processing; however, the specific pathways underlying locomotor deficits and the cerebellar circuits that are disrupted in DS remain poorly understood. Defining specific abnormalities in motor behavior and identifying the brain regions and neurons which are functionally involved will provide the basis for developing potential therapies for treating motor problems in people with DS. The main goal of our studies is to identify specific alterations in the circuitry of the cerebellum that result in locomotor dysfunction in DS.
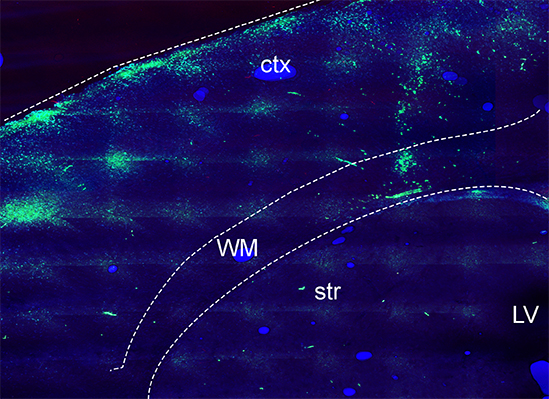
White Matter Oligodendrocytes from trisomic TSDn65 mice. The image above shows oligodendrocyte progenitor cells (OPCs) from TsDn65 brains labeled with a green cell tracker and transplanted into the white matter of homozygous shiverer mice (Mbp shi-/-) at postnatal day 5. At 10 days after transplantation, trisomic OPCs migrated from the site of injection to the cerebral cortex but did not generate the same number of mature oligodendrocytes as wild-type OPCs. Ctx: cortex; WM: white matter; str:striatum; LV: lateral ventricle.